四、怎样提高阳性率(节选自《遗传病精准诊断攻略》)
- 阳性率低,是什么原因?
在展开讨论之前,我们先要正确理解阳性率的概念。阳性率是个统计数字,直接受患者入组选择标准的影响。遗传病患者只是全部人群的一小部分。对全部感冒发烧的成年患者做单基因病的基因检测,肯定不会有高的阳性率。对于遗传因素只占一小部分的疾病,本来就不应该把基因检测的高阳性率作为追求目标。
但对于遗传占主要致病因素的疾病,我们就应该发展应用更先进的技术和诊断模式,去达成更高的阳性诊断率。本节所讨论的阳性率,均为对遗传占主要致病因素的疾病而言,这是本节全部讨论分析的前提。
想当年用Sanger测序诊断遗传病的时候,医生从厚厚的基因项目册中挑选基因进行检测。如果真的检出致病突变了,医生会欢呼“中奖了”,可见当时的阳性率是多么低。
尽管现在有了NGS(高通量测序),一次可以把某种临床症状相关的几十或几百个基因打个Panel来进行检测,但真正的致病突变的检出率仍然仅仅在10~20%之间。那么究竟有哪些因素导致阳性检出率低呢?
检测范围的局限1——漏检疾病
其实阳性率,和检测的范围,或者说检测策略是直接相关的。小Panel会存在打包不全的问题,甚至在对于复杂疾病有可能不知道选择哪个Panel,在这一点上,很明显全外显子要大大优于Panel策略。可以说,小Panel,是从低通量时代(Sanger测序时代)到高通量测序时代(全外、全基因组测序)的过渡期的产物。
涉及神经、心脏、肝脏、骨骼肌、肾脏等能量需求较高器官异常的疾病,不能排除线粒体病。漏检mtDNA和线粒体核基因是非常常见的现象。
有些皮肤病、血液病是体细胞突变所致,只有采集病变组织检测,才有可能得到阳性结果,只检测白细胞,按孟德尔遗传病去分析,一般不会得到阳性结果。
漏检变中、大型变异形式
一般小Panel只检测点突变。很多情况医生没有意识到有可能是中型(外显子缺失重复)、大型变异(CNV)所致,因为医生所熟知的、临床上能辨识的涉及到中型、大型变异的疾病很少。然而漏检中、大型变异,也会导致明显的检出率下降。
只检测先证者,没检测家系(Trios)
除了检测范围之外,要想提高阳性率,在检测策略选择时还应该尤其注重家系(Trios)策略,也就是患者(先证者)与其父母同时进行NGS检测。普通的先证者“单人再验证”检测策略很有可能忽略掉生物学和临床特征不典型的基因变异,但家系筛查可以借助遗传学共分离分析,筛选出致病突变。有研究表明,对于各类疾病,家系(Trios)策略可以使临床外显子组的阳性率获得40%~300%的不同幅度的提升。
基因数据分析能力的局限
既然阳性率低是因为检测范围不全,那么仅仅放大检测范围就可以解决阳性率问题吗?还不是那么简单。检测出来了,但如果数据分析跟不上,仍然会降低检出率。因为检测范围扩大,数据分析的难度也就同步加大。全外显子组检测出来的变异数量,是小Panel的上百倍,数据解读的压力也陡增百倍。
临床特征收集及分析不到位
前文已述,即便基因检测技术和检测策略都没问题,即便运用数据分析工具,如果最基础的临床表型数据不全面不准确,标准化不到位,都有可能对分析的准确性大打折扣。这需要有强大的临床表型录入和管理工具,更具要医生建立正确的意识,重视表型收集,认真使用工具进行录入。
下面我们就上述这五个导致阳性率低的原因,逐一展开探讨。
- 提高阳性率之——检测范围越全越好
检测范围越全,越能避免漏检
检测范围越全,越能避免漏检,这个道理很容易懂。
目前遗传病NGS检测策略主要有小Panel、临床外显子、全外显子、CNVseq几种策略,低阳性率往往和检测策略选择有关。
Panel策略,也叫疾病相关靶向测序策略,是对某种临床症状相关的几十或几百个基因打个包。
相比于全外显子,Panel策略的优势在于成本较低,数据分析难度也小,对于相对特异的疾病群,经验丰富的医生运用起来效果也不错。
但从各种遗传病诊断的总体实际效果来看,Panel策略的阳性率只有10~20%,为啥这么低?原因有三:
一是打包不全,谁也不能保证能打全,即便是穷尽了已知的致病基因,每年仍然会发现几百种疾病和相关基因;
二是Panel一般是基于某个系统的某种异常的临床特征来打包的,只是涵盖了这种临床特征相关的基因,但当遇到多系统疾病的时候,尤其是表型不典型的时候,即便相关基因在Panel里面,有可能医生会下错申请单,去检测另外的Panel了;
三是当一个人真的是有两种不同系统的疾病的时候,仅针对任何一个单一系统的Panel都不可能覆盖全。
相比之下,全外显子组(人类全部2万个基因),能较好的解决打包不全的问题,阳性率大约可提升至35~45% 。
2015年一篇关于儿童神经发育疾病的研究揭示,做Panel(小包)检测不出来的119例患者,用全外显子(大包)可以额外增加24%的阳性率。
对于难以诊断的疑难表型,尤其是涉及多系统疾病,或者是基因异质性突出,比如(但不限于)多系统疾病、各种畸形自闭症、智障、癫痫、发育迟缓、贫血、矮小等,涉及数百上千个致病基因,用全外显子会比Panel有较为明显的优势。
前面我们讲了检测范围越全面,就越有可能避免漏检,就越能提高阳性检出率。另外检测范围追求越全越好的意义还不仅于此。
检测范围越全,诊断越严谨
在低通量时代,因为技术局限性,测得太多成本就太高,所以临床上一般是先怀疑(说通俗一点就是猜)哪种病,就检测哪个基因,倘若在这个基因上还真就找到了一个变异的话,就很容易先入为主、对号入座,确诊是这种病了。
这种情况就类似盲人摸象,摸到的只是局部,摸到象腿就说大象是柱子,摸到尾巴就说大象是绳子,检测到这个基因有变异就说患者就是这个病,难免以偏概全。
其实,这种做针对性的检测——单个基因的一代测序,或是小Panel,大多是低通量时代的思维的延续,这种低通量思维主要基于以下的逻辑前提:
- 单基因病的表型非常特异,不会存在多种疾病混淆的情况,所以怀疑哪种病就做这种病的基因就OK。
- 基因变异的致病性很明确,在目标基因找到变异就可以确诊了。
然而实际的情况可能恰恰相反:
- 疾病的复杂性远超预期:单基因病种类繁多,特征相似的疾病往往很难鉴别诊断,对某种疾病的预先诊断,很有可能是错误的。
- 变异致病性的不确定性远超预期:与患者表型相似的疾病可能有很多,意味着有可能在很多个基因上都存在变异,但并不是所有的变异的致病性都很明确,需要在众多变异中进行选择。
在过去的低通量时代,没有办法把所有疾病进行全面筛查,我们只能是预先诊断,再做有确证性的检查,尽管不严谨,但也只能如此。但如今进入高通量测序时代,我们就有了一窥全貌的机会,我们完全可以在更广阔、全面的遗传背景上去确证,在我们怀疑的这种疾病之外,没有其他更值得怀疑的疾病的选项了。
因此高通量时代的思维应该是,先不做预判(即便有预判也先不急下结论),先测,尽可能全面检测,比如全外显子以及全基因组CNV,测完再从整体全貌上把握最像哪种疾病,或许全面测完之后才发现有比原来预想的更吻合患者特征的疾病,和更具备致病性的基因变异。
当然,即便是做全面的检测,也存在漏检的可能,但这总比只测一个基因或者小Panel要全面得得多,严谨得多。
因此在检测策略上,能做全外显子+CNV,就尽量不要选择小Panel。要注意的是,做这么大范围的检测,需要选择Trios家系策略去借助共分离筛选变异,否则数据分析难度太大,也难以发现新发变异。
别忘了检测线粒体基因组(mtDNA)和线粒体核基因
除了上述因素之外,漏检另一套基因组——线粒体基因组(mtDNA)也是影响阳性率的因素之一。在线粒体病患者中,mtDNA致病突变的检出率大约为10~20% 。
教科书上常列举一些线粒体病综合征(如Leigh、KSS、MELAS、MERRF、MNGIE等),这反而会给人以误导,认为只有这些综合征才是线粒体病,其实线粒体病的表现远不止这些综合征。线粒体病未必都是多系统受累的,很多单一系统特征的疾病都有可能是线粒体病所致,如单纯的癫痫、自闭症、心肌病、肾病、肝病、肌病、身材矮小等。
有些Panel策略没有包含线粒体核基因,更不包含mtDNA,在选择用什么Panel上就会大伤脑筋。其实如果选择全外显子组,就一定包含线粒体核基因,再加测mtDNA,就能把线粒体病机制全面考虑进来。
- 提高阳性率之——大中小变异一网打尽
疾病表现与基因变异形式没有通用相关性
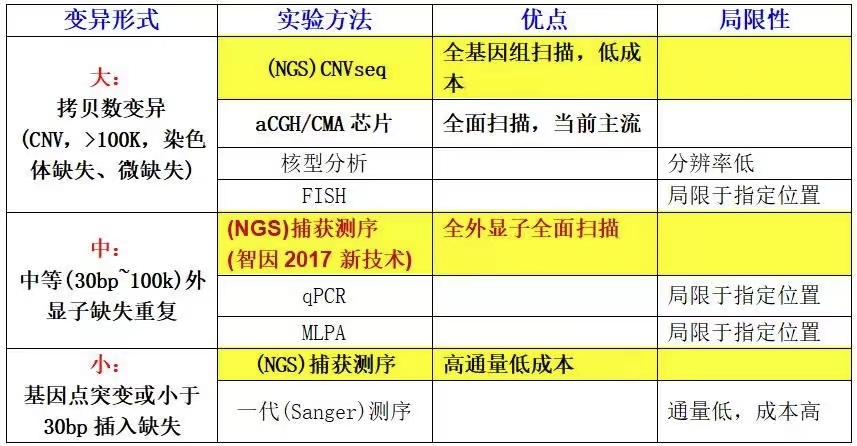
单基因病,主要是由于特定的一个基因的功能异常所致。至于这个基因是怎样异常的,是点突变(小)、外显子缺失(中),还是CNV(大)导致的这基因的异常,应该说这三种都有可能,而且都有可能导致相同或相似的疾病表现。
因此诊断遗传病选择检测策略,应该同时涵盖大中小三种变异,一个都不能少。
不要根据疾病表现去选择推断变异形式,比如多发畸形并非CNV的专利,如果点突变发生在多系统综合征的基因上,照样会导致多发畸形;单一表型也不是点突变的专利,如果CNV覆盖了单一表型的疾病的相关剂量敏感基因,导致整个基因拷贝数异常,也有可能会同样出现该疾病的单一表型特征。
中型变异的致病概率很可能不亚于点突变
中型变异(外显子缺失重复),是非常容易被忽略的致病变异形式。实际上,中型变异很可能在致病概率上不亚于点突变。
我们所熟知的相对常见的遗传病,无论是DMD(进行性肌营养不良)、SMA(脊肌萎缩症)、CAH(21羟化酶相关肾上腺皮质增生症)等,都有接近甚至超过一半的病因是中型变异引起。难道只有这几个基因集中体现为中型变异,而其他四千多个遗传病的基因都只体现为点突变?目前没有理论能支持这种判断。但值得深思的是,凡是常见的遗传病基因都体现出很高的中型变异致病概率,仅凭简单推断可知,是因为常见,所以研究得就透彻,小型、中型变异都有人研究,就发现了中型变异的高致病概率,那么其他基因不排除也是同样的,中型变异具有高致病概率!
为什么中型变异会有如此高的致病概率?一方面是因为中型变异很常见,另一方面,是因为中型变异有很强的生物学危害性,绝代多数都是强致病性变异。
传统的实验方法(如MLPA等)只能对中型变异进行确证式的检测,所以也只能对常见的几种遗传病进行分析。
在过去,NGS的捕获测序,无论是小Panel,还是全外显子,都无法解决中型变异的问题。
2017年智因东方(原德易东方)推出的全外显子测序的中型变异分析技术解决了这一难题,该技术可以对全外显子数据进行全面筛查,对2个以上的外显子缺失重复的准确性高于90% 。因此,智因的技术突破实现了一次全外显子测序同时检测小型和中型两类变异,增强了检测分析的性价比。
大型变异CNV也是不可忽略的检测目标
全基因组CNVseq是解决CNV检测的适用策略。过去几年CNV检测主要是依赖于染色体芯片(CMA或aCGH),当前基于全基因组二代测序的CNVseq有着替代染色体芯片的潜质。从技术准确性上看,目前CNVseq可以检出100k以上的缺失重复,准确性高达99%以上,与染色体芯片难分伯仲。CNVseq的优势在于检测范围更全,可以全基因组几乎没有死角的检测(基因组重复区域分析仍然是需要解决的难题),而且价格上比染色体芯片更具优势。染色体芯片的优势在于其技术成熟度,在遗传病诊断领域应用时间相对比较长,CNVseq类似技术过去主要应用在NIPT(孕妇外周血对胎儿筛查唐氏综合症等染色体整倍体异常),这是染色体芯片的敏感度难以触及的领域。可以期待CNVseq未来在遗传病诊断领域的成为主流CNV检测技术。
CNVseq可以解决儿童遗传病的约20%的阳性率,对于新生儿或产前疾病则阳性率更高。
那为什么临床上会忽略CNV呢?这可能和一个认识误区有关,即认为只有出现多发畸形才会考虑CNV,否则就只考虑点突变。其实临床表型(是否有多发畸形)与突变形式(点突变还是CNV)之间,没有必然联系,点突变可以导致多发畸形,CNV可以仅体现单一特征。因此要想避免低阳性率,在申请检测策略上,应该是点突变和CNV的检测,一个都不能少。
大中小变异,2017智因NGS涵盖
- 提高阳性率之——家系(Trios)模式
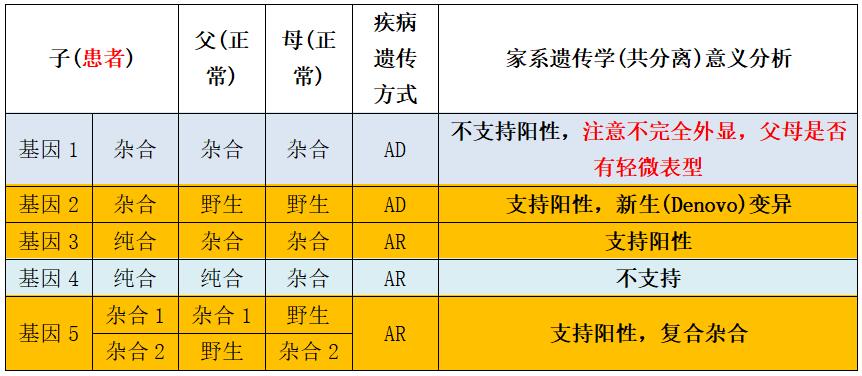
致病突变的必要条件——家系遗传共分离
如果一个变异是导致某种单基因病的,那么患者一定是以致病方式携带(比如对于常染色体显性遗传病,就是杂合携带,对于常染色体隐性遗传病,就是纯合或复合杂合方式携带),而其没有患病的家族成员(比如其父母)则应该是以非致病方式携带(比如对于常染色体显性遗传病,就是不携带的野生型,而对于常染色体隐性遗传病,就是杂合携带或不携带)。这种在一个家系里,相同表型和基因型的连锁绑定,以及不同表型和基因型的明确区分,被称为遗传学共分离。
遗传学共分离现象为判断变异的致病性提供了重要依据。
家系(Trios)模式为共分离分析提供了可能
传统的遗传病基因检测,由于成本原因,大多是先检测先证者的编码区序列,如果发现了可疑的变异,再用Sanger测序验证其父母是否携带该变异,进而判断是否存在遗传共分离,这可以叫做“单人再验证”模式。
家系(Trios)模式则是一次同时对先证者及其父母进行NGS检测,对先证者的每一个变异都进行三人的平行比对,看是否符合共分离。
非标准(超过三人)的家系模式分析
其实家系检测分析不限于标准家系——患儿加父母一家三口(Trios),非标准家系更好,非标准家系是指在此基础上再增加其他家庭成员,参与检测的家系患者成员越多越好,患病成员涉及代际越多越好,患者的无表型兄弟姐妹越多越好。
家系(Trios)模式的优越性
与传统的先证者检测相比,家系(Trios)模式可以:
降低假阳性率
在数据分析时最头疼的是在患者的数据中,有大量生物学和临床特征似是而非。难以判断致病性的变异,借助家系(Trios)模式,可以干净利索的过滤掉不符合共分离原则的变异,瞬间感觉整个世界都清净了!
提高真阳性率
有些变异可能是生物学和临床特征不那么典型支持致病的,有可能在数据分析时被过滤掉。但是借助家系(Trios)模式,可以筛选出符合共分离原则的变异,尤其是新发(Denovo)变异,这类变异在ACMG标准中可是强烈提示致病的级别!
2014年Lee H在一组800多例临床外显子组测序的研究对比中发现,家系(Trios)策略可以使临床外显子组的阳性率从22%提升至31%,甚至在发育迟缓类患者中,仅测单人的阳性率仅有9%,家系(Trios)模式高达41%!
缩短诊断周期
如果没有家系(Trios)数据,则有大量似是而非的变异需要人工分析,不仅如此,人工分析后还要用Sanger测序法去验证是否符合共分离。这对于全外显子组或全基因组来说,这个工作量是恐怖的,周期也是不确定的,一般会拖延两周以上,而且很有可能白费了半天劲,仍然漏掉了致病突变。但借助家系(Trios)模式,则可一目了然挑选出共分离的变异。
降低检测费用
看到这个说法,不少读者自然会有疑问,家系(Trios)检测三个人,怎么会比检测单人反而降低费用呢?因为如果没有家系(Trios)数据,则有大量似是而非的变异要用Sanger测序法去验证是否符合共分离。这个费用是不菲的。按国内大多数地区的基因检测收费标准来计算,一个位点一个人500元,家系三人就是1500元,如果要验证10个位点就要1万5千元!但借助家系(Trios)模式,可直接精准找到致病突变,则可大幅降低患者的检测费用。
家系(Trios)模式有这么多好处,说它带来了遗传病诊断的革命也不为过。
- 提高阳性率之——可信赖的数据分析
前面讲要提高阳性率,就要尽可能的扩大检测范围,并且要做家系Trios策略。但检测出来是相对容易的,检测出来的海量数据能否分析出来则是相对难得多、技术含量高得多的挑战。要选择可信赖的数据分析平台或机构。
怎样的数据分析才是可信赖的呢?
涵盖三要素
生物+遗传+临床,三要素同时支持致病性判断,这是一个变异的致病性的充分必要条件。数据分析平台应基于这三要素,对海量变异进行分级筛选。(详见:《详解:关于遗传病精准诊断阳性率的探讨》)
生物学要有五级致病性分析
分析程序兼容大(CNV)、中(外显子缺失重复)、小(点突变)三种类型。
对已报道变异,要能支持海量文献数据库的索引。对未曾报道的变异,要能提供变异对蛋白结构的预测性分析,而且要基于不止一个软件。
要基于多个公共数据库、尤其是中国人种的万人级别以上的数据库的MAF值,去过滤无害变异。
要兼容ACMG变异评级标准中的分析内容,包括保守性分析等。
遗传学要有共分离分析
要基于家系Trios模式,进行遗传共分离分析。
临床表型分析要基于结构化数据库
要基于结构化的人类七千种遗传病的临床表型数据库,结合患者的临床特征,进行匹配分析。
要有自动综合评级
要能提供结合三要素分析之后的自动综合评级结果,这样才能减轻人工分析的压力。
数据质量、分析依据要透明展现于在线平台
数据分析的逻辑和过程,以及综合形成的分析判断依据,都应展示于在线平台上,可供医生查看。
要可参与
在线平台能支持医生参与分析,提供方便的筛选、排序等功能,提供临床特征的在线修改、即时匹配。
要基于中国人的遗传病患者大数据
分析系统应基于中国人的遗传病患者的全外显子家系的大数据,这样才有可能结合大数据进行精准诊断。
- 提高阳性率之——临床特征采集和标准化录入
临床特征标准化是临床和科研分析的关键
在临床诊断应用中,临床特征标准化是NGS检测分析前的临床特征收集工作最关键的“临门一脚”。否则病历信息收集得再全面也体现不出效果,因为病历描述很可能计算机不识别。
在科研分析中,要对大量样本形成的大数据进行挖掘,如果临床表型的标准化做的不好,就很难准确挖掘其大数据中的科研价值。比如我们要分析合并“癫痫”与“贫血”两种表型的患者,共同在哪个或哪些基因上产生变异,从而发现新的致病基因,就需要这些数据都有很好的临床特征的标准化管理。否则如果最初开单录病历表型词的时候就不规范,后面再做科研时,基础数据不完善导致难以挖掘大数据价值,就可能悔之晚矣。
如何准确详实地收集患者临床特征
越详尽越好
包括症状、体征、实验室检查、辅助检查、既往病史、家族病史等;
出现多系统特征的,不要漏掉某些系统异常,而误以为是单系统疾病;
能细致就不要粗劣,比如多指畸形,就不要说成笼统的手足畸形;
有家族史的,其家系成员有不典型或轻微表型就不要当做正常无表型。
容易被忽视的
外貌特征(外貌特征及形体特征可向上概括为查体异常):
头面部:头围,注意小头畸形,发际,额部异常,眉毛稀密及眉弓,耳的大小,耳位高低,眼距,眼裂,睫毛,鼻翼发育,鼻根,人中,有无唇裂、腭裂和高腭弓,口腔内注意舌体及牙齿情况,小下颌畸形,毛发稀疏和毛发颜色。
形体特征:
注意上身长与下身长的比例、指距、手指长度、多指(趾)并指(趾)、皮肤和毛发色素、手纹、外生殖器等。注意黄疸、肝脾大和神经系统症状、一些不正常的汗味或尿味等。
实验室及辅助检查:
血、尿、便三大常规,血生化、免疫学、凝血功能、自身抗体、病原学等。代谢病需要提供血串联质谱分析结果及尿气象色谱分析结果。针对具体案例,需要提供具有重大诊断价值的相关检查结果。
不同科室或不同系统的遗传病侧重关注检查方向有所不同,应搜集该专科的专科查体及专科检查资料。例如眼科(视力、眼底、视网膜电图);肾内及泌尿(尿常规、电解质、酸碱平衡、超声等影像学等);血液科(血常规、涂片、骨穿等);神经系统癫痫(临床发作特点、脑电图、MRI等)、神经肌肉病(肌力、肌张力、肌电图、肌肉活检)。
阴性特征
阳性(异常)特征固然重要,但阴性特征也很重要,可以用来排除某些疾病。
祖孙三代家族病史,家系成员的轻微表型
这个容易被忽视,也是特别重要的信息。如果家系成员的表型信息缺失,或者存在错误,遗传学共分离分析就无法完成,或者完成的也是错误的。问询家族病史的时候,涉及的代际和成员越多越好。另外特别注意家系成员的与先证者疾病类似的轻微表型,因为这代表着该家系成员是携带者,对于AD遗传病,则是不完全外显的患者,对于AR遗传病则是杂合携带者。
比如有位协和的遗传学老教授介绍过的一个病例,患儿罹患视网膜母细胞瘤,检测到患儿携带杂合的疑似致病突变,患儿母亲也携带但不发病。医生觉得奇怪,便叫来患儿母亲仔细观察其眼部,发现其眼底有白斑。这就是一个典型的不完全外显的例子,患儿母亲的确没有发病,但是有轻微表型。如果不是医生的较真,认为母亲是无表型,按遗传学共分离分析,就有可能把这个突变当做不致病变异给过滤掉了。
如何做好临床特征标准化录入?
用标准化医学术语,统一临床特征的描述
如果要实现让计算机自动匹配患者疾病与七千种遗传病的特征的相似性,就需要让病历信息成为计算机可识别的元素,这就需要让临床特征的描述标准化,也就是运用标准化医学术语,统一临床特征的描述。
HPO(Human Phenotype Ontology,人类表型本体论),是迄今为止关于疾病(尤其是单基因遗传病)特征的结构化管理的数据库。它把七千多种遗传病的海量表型特征,进行结构化处理,去掉重复后得到一万多种表型词。这些表型词之间有层级逻辑关联。
利用全谱云平台进行临床特征标准化
国内最早完成了HPO的汉化的机构是智因东方(原德易东方)——早在2015年初就联合各临床学科的专家团队率先完成,并把汉化版HPO嵌入全谱精准诊断云平台系统中。
比如癫痫发作这样一个特征,相同或相似的意思,可以有很多种叫法,如癫痫发作、抽羊角风等等,在HPO数据库中则统一为“痫性发作”这样一个标准称谓。
另一方面,在HPO收录的七千多种遗传病的临床特征描述,其实都是HPO中的表型词的组合,当医生在录入患者表型词时,如果选择标准词“痫性发作”,就可以与涉及“痫性发作”几百种疾病的表型相匹配。
如果涉及到关于“痫性发作”更细节的描述,则可在子特征(如癫痫持续状态、全身性发作、高热惊厥、局灶性痫性发作、癫痫性痉挛、症状性发作、意识障碍发作等)以及更下一级特征中去选择。

模糊检索
可以输入简单的关键词,在HPO标准词库中查找与患者某特征最贴近的表型词。对于经过专业训练的医生,一分钟内完成一个患者的表型的标准化录入是完全有可能的。
单选最准确的层级
在模糊查询的众多表型词中,单选最相近的表型词,不要把像与不像的都一股脑选上,否则会造成匹配评分的偏差。